 供需大厅
供需大厅
 登录/注册
登录/注册 供应商登录
供应商登录










荣格工业资源APP
了解工业圈,从荣格工业资源APP开始。
欢迎来到荣格工业资源网!

供需大厅

登录/注册
公众号

更多资讯,关注微信公众号
小秘书

更多资讯,关注荣格小秘书
邮箱
您可以联系我们 info@ringiertrade.com

电话
您可以拨打热线
+86-21 6289-5533 x 269
建议或意见
+86-20 2885 5256
顶部

荣格工业资源APP
了解工业圈,从荣格工业资源APP开始。



在生物医药的微观丛林中,外源性纳米材料一旦进入人体循环,便会立即陷入蛋白质的「重重包围」。这种现象,往往决定了纳米药物的最终命运:是被免疫系统当成异物清除,还是能精准敲开肿瘤细胞的大门?
长期以来,研究人员一直试图预测蛋白质与纳米颗粒之间的这种「相亲」过程。
然而,面对由 20 种氨基酸排列组合而成的海量蛋白质序列,传统的实验观察和计算模拟显得捉襟见肘。
是否能找到一套通用的「化学密码」,在计算机上预判两者的结合倾向?
近日,《国际医药商情》获悉,芬兰于韦斯屈莱大学(University of Jyväskylä)纳米科学中心的研究团队在 Aggregate 期刊发表了一项突破性进展。

他们利用机器学习构建了一套计算模型,成功破译了配体保护金纳米簇(Gold Nanoclusters,AuNCs)与蛋白质相互作用的化学规则,为生物成像和靶向给药提供了新参考思路。
Part 1
算力瓶颈
在纳米材料的世界里,金纳米簇因其天然的荧光特性、优异的生物相容性以及可通过肾脏排泄的安全性,成为了生物成像和生物传感领域的「明星」。
为了使其在体内稳定存在,科学家通常会在金核表面包裹一层有机配体,例如对巯基苯甲酸(p-MBA)。
然而,当这些被保护的金纳米簇与蛋白质相遇时,界面上的化学反应变得极其复杂。
蛋白质由 20 种氨基酸组成,这些氨基酸通过肽键连接形成特定序列。当蛋白质接触金纳米簇表面时,其构象会在水环境驱动下发生改变,氨基酸残基与纳米簇配体层之间产生复杂的吸引与排斥作用。
过去,行业内普遍采用分子动力学(MD)模拟来观察这种动态过程。但是,计算成本是一个绕不开的「金钱黑洞」。
对于含有两个氨基酸的二肽,排列组合有 400 种;三个氨基酸的三肽有 8000 种;一旦增加到四个或更多,组合数量便呈指数级爆炸。即便调用最顶级的超级计算机,也无法穷举所有可能的交互场景。
「尽管科学界为理解生物分子与纳米颗粒界面的结构关系付出了大量努力,但建立这些相互作用模型的研究仍然匮乏。」研究通讯作者、于韦斯屈莱大学教授 Hannu Häkkinen 指出,「开发这样的模型可使高通量计算筛选成为可能,无需执行大批量模拟即可识别出具有应用价值的体系。」
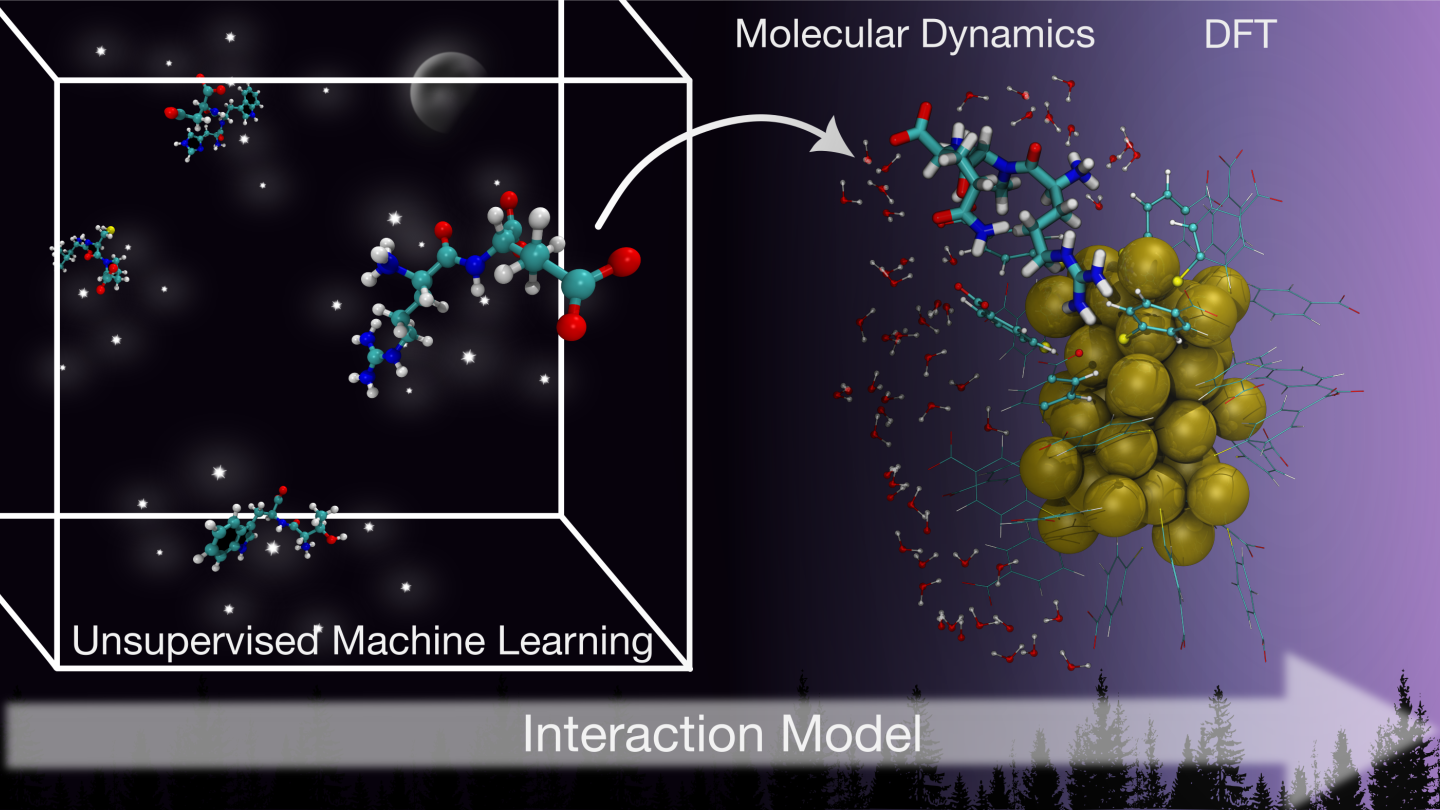
Part 2
寻找「通用模版」
于韦斯屈莱大学的研究团队并未走传统的「一事一议」路线,而是试图建立一个具有通用性(Generalisable)的模型。他们将目光锁定在了 Au₃₈(p-MBA)₂₄ 这一经典模型上,其金核结构已被充分表征,p-MBA(对巯基苯甲酸)配体提供水溶性,适用于生物医学场景。
研究人员首先利用非监督机器学习中的聚类分析(Clustering Analysis),对氨基酸和短肽进行了化学特征提取。
核心思路是,不直接依赖纳米簇结构或溶剂化效应,而是定义与相互作用相关的化学性质。研究团队选取「酸性基团数」和「碱性基团数」作为目标性质,利用分子指纹描述肽段化学结构,通过成对相关性分析筛选与目标性质高度相关的特征变量。
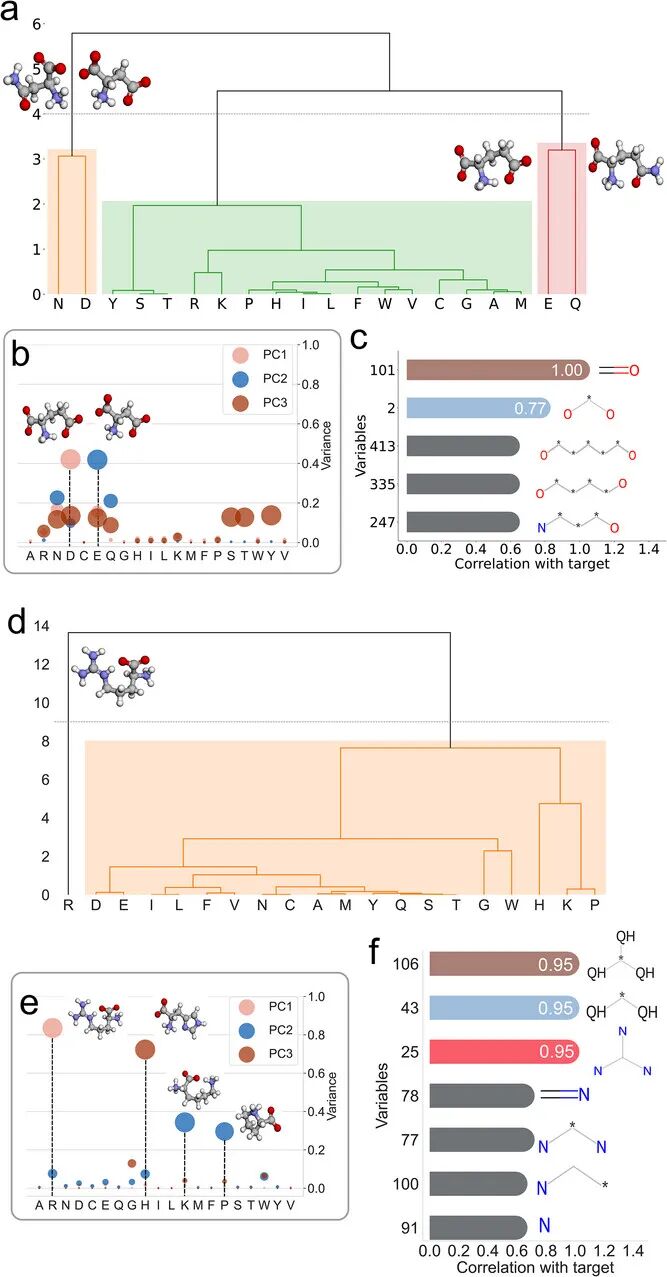
图:分级聚类树状图(Dendrogram),直观呈现不同氨基酸在酸碱属性上的归类逻辑,特别是精氨酸(R)与天冬氨酸(D)、谷氨酸(E)的显著差异
聚类分析在四个子集上进行:双肽的酸性/碱性相关性,以及三肽的酸性/碱性相关性。单氨基酸的分析用于理解各氨基酸的独立贡献。
聚类分析揭示了清晰的化学规律。
对于酸性基团,天冬氨酸(D)和谷氨酸(E)的贡献最为显著。这两种氨基酸侧链含有羧基,与目标性质的相关性最强。天冬酰胺(N)和谷氨酰胺(Q)虽然结构相似,但侧链为酰胺基而非羧基,相关性明显较低。
对于碱性基团,精氨酸(R)的贡献占据主导地位,其胍基对主成分 PC1 的贡献达到 84%。组氨酸(H)、赖氨酸(K)和脯氨酸(P)次之,甘氨酸(G)和色氨酸(W)也显示出一定相关性。
这一结果与 Au₃₈(p-MBA)₂₄ 的化学结构高度吻合:p-MBA 配体通过硫原子与金核结合,羧基朝外构成纳米簇表面。在 pH 7 条件下,羧基去质子化使表面呈强负电性。碱性基团接受质子的能力使其更易与负电表面形成有利相互作用。
Part 3
精氨酸的「胶水」效应
当研究从单个氨基酸扩展到二肽和三肽时,机器学习模型给出了更加具体的预测。
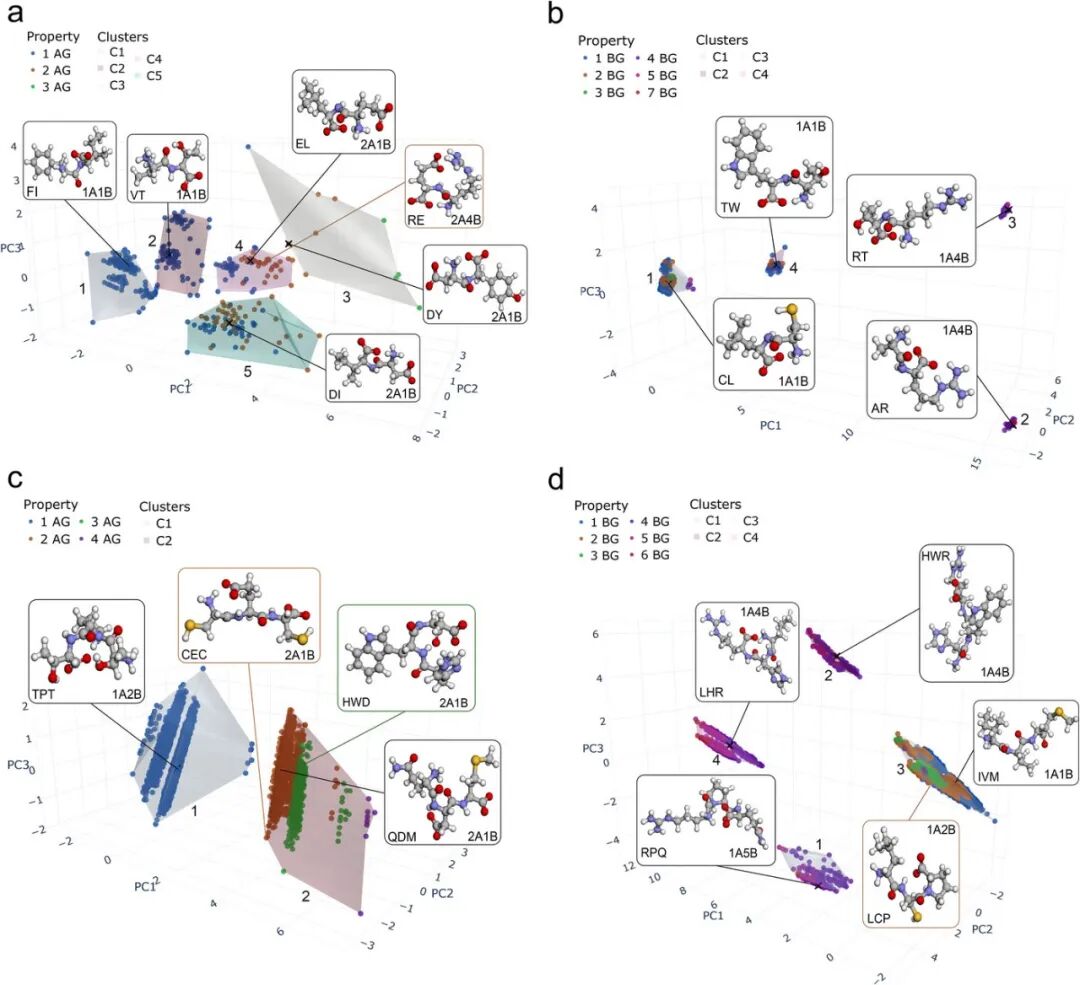
图:K-means 聚类的散点图,通过不同的颜色区域显示肽段结合能力的强弱分布,并标注出作为验证对象的代表性肽段
研究发现,含有精氨酸(R)的肽段几乎总是位于结合能力的顶端。
这背后的化学逻辑在于,金纳米簇表面的 p-MBA 配体在 pH 7 的生理环境下带负电,而精氨酸侧链的胍基(Guanidine Group)不仅带正电,还具有极强的质子接纳能力,能够像「钩子」一样深扎进配体层的缝隙中,直接与金核界面的硫原子发生作用。
相比之下,那些富含酸性基团的肽段(如 EL、CEC)则表现得极为冷淡。模拟数据显示,这些肽段在水溶液中往往只是在纳米簇周围「漂浮」,很难建立稳定的物理接触。
为了验证这些预测,研究人员动用了位于芬兰的 LUMI 超级计算机,进行了长达数百纳秒的 MD 模拟和高精度的密度泛函理论(DFT)计算。实验观测到的肽段结合顺序(RPQ > LCP > HWD > CEC)与机器学习模型的预测完全吻合。
Part 4
加速下一代纳米药物开发
此项研究的意义远不止于破译了几条化学规则。对于制药行业而言,它提供了一种「高通量筛选」的计算方案。
在传统的药物研发流程中,设计一种新型的靶向纳米造影剂可能需要数月的实验摸索。而现在,通过这套机器学习框架,研究人员可以在几秒钟内预判某种蛋白质受体是否会与特定的金纳米簇发生强相互作用。
「我们的目标是构建一个不局限于单一系统的模型。」博士后研究员 Brenda Ferrari 表示,「虽然目前还存在局限性,但我们已经拥有了一个可以扩展到广泛蛋白质-金纳米簇交互研究的基础工具。」
研究相关代码已开源,包括肽段生成工具 GenPep、PDB 文件生成插件 MoltoPDB 和完整分析流程 UML-for-peptides。
在 AI 浪潮席卷全球制药业的背景下,纳米材料学与计算科学的深度融合已是大势所趋。于韦斯屈莱大学的这项工作,实际上是为纳米医药的底层设计逻辑打上了一个坚实的补丁。
未来,随着模型引入更多关于极化率、电子效应以及环境溶剂化效应的参数,有望看更多具备预测物质相互作用能力的模型,可在无需大量物理化学模拟的情况下实现高通量筛选,有望加速生物成像、生物传感和靶向药物递送领域的发现进程。
参考资料:
Aggregate (2025): e70213, DOI: 10.1002/agt2.70213
来源:国际医药商情
作者:John Xie


